Study Quantifies Limitations in Using Marker Genes to Predict Microbial Capabilities
– SEPTEMBER 17, 2019
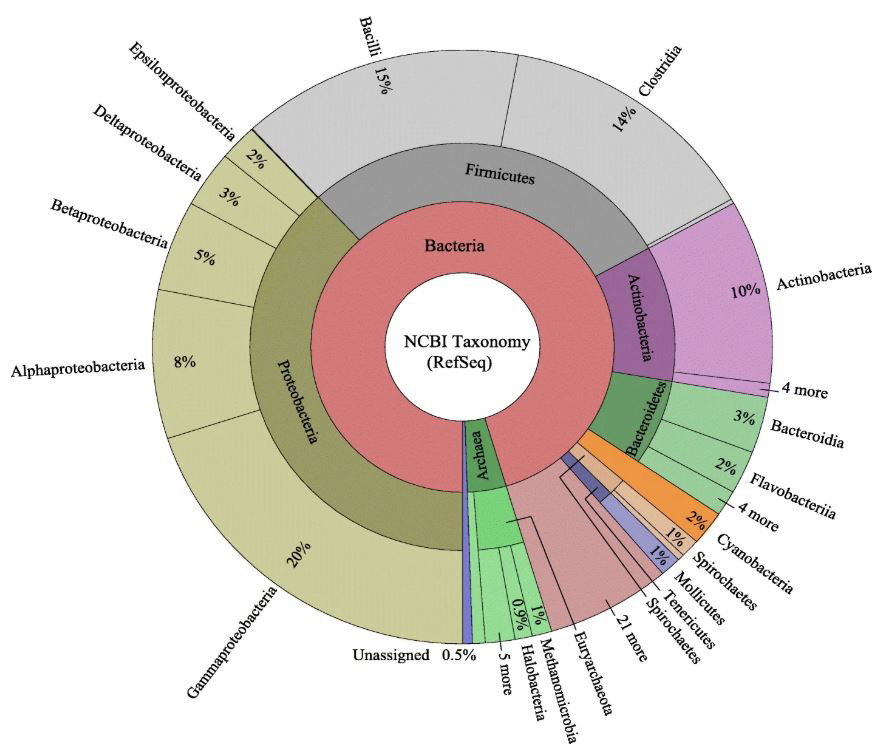 This KronaTool map depicts the relative taxonomic breakdown of the genomes used in this study Sevigny et al., BMC Genomics volume 20, Article number: 268 (2019). The inner circle represents genomes at the domain, the middle circle corresponds to phylum, and the outer circle represents data at the class level. Figure 2 in the study, used with permission from Joseph Sevigny. |
Scientists conducted the largest comparison to-date of publicly available sequenced bacterial genomes to provide the first in-depth look at using high-throughput marker genes to profile a microbial community’s functional capability. Using the relationship between marker gene identity and shared protein-coding gene content, the researchers found that even phylogenetically identical organisms did not share substantial portions of their gene products. Genomes shared on average less than 75% of their protein-coding gene content, regardless of the marker gene(s) used, leaving about a quarter of any given bacterial genome as unpredictable and dependent on a specific microbial niche or environmental conditions. The authors suggest that researchers use whole-genome/metagenomic sequencing to backup findings of inferred microbial functional capacity based on marker gene phylogeny, typically used in amplicon-based studies.
The researchers published their findings in BMC Genomics: Marker genes as predictors of shared genomic function.
There has been increased scientific interest in predicting what microbial communities can do, such as identifying oil-degrading marine microbes following Deepwater Horizon, based on phylogenetic identification inferred from marker genes. However, phylogeny is largely dependent on database coverage, which may lead to missing novel functions or overestimating functional capacity. Phylogeny also does not consider inputs that help shape microbial functions like local environmental conditions, taxa abundance, and phage presence.
“Metabarcoding (amplicon or marker gene-based sequencing) studies are applied to analyze environmental samples and determine what bacteria (or other microbes) are there,” explained study author Joseph Sevigny. “The idea is to sequence a single or partial marker gene sequence for each organism present in the sample and subsequently match these sequences to a database of known species. These types of studies provide valuable measures of phylogenetic diversity and the relative abundance of different taxa within or across samples, but they do not provide direct information about what those bacteria can do. This study aims to test how well you can predict the functions of a given bacterial community with marker gene-based studies.”
The team analyzed 4,872 well-annotated representative prokaryotic genomes from the National Center for Biotechnology Information. Most gene ontology terms were shared across the genome dataset, such as DNA repair and binding; however, observations showed that important and unique functions were significantly more present in the novel/unshared gene set. For example, transposase activity (molecular function), transposition (biological processes), and vesicle membrane (cellular component) were highly present in the novel dataset. Functions related to metabolic processes, such as glucosidase activity or fucose metabolic processes, were found in the unshared dataset.
The authors suggested that the introduction of non-native DNA from events like horizontal gene transfer and gene deletion/loss of function may account for their observations. For example, lineages like E. coli and Vibrio laterally transfer DNA, which could result in a large pool of genes that are unshared between phylogenetically related organisms. They concluded that grouping microbial species based on marker gene(s) sequence similarity and predicting functional content likely leads to overestimating functional capacity and missing novel functions, an aspect that may motivate such studies in the first place.
Data are publicly available through the Gulf of Mexico Research Initiative Information and Data Cooperative (GRIIDC) at R5.x272.000:
The study’s authors are Joseph L. Sevigny, Derek Rothenheber, Krystalle Sharlyn Diaz, Ying Zhang, Kristin Agustsson, R. Daniel Bergeron, and W. Kelley Thomas.
By Nilde Maggie Dannreuther. Contact maggied@ngi.msstate.edu with questions or comments.
************
This research was made possible in part by a grant from the Gulf of Mexico Research Initiative (GoMRI) to the University of New Hampshire Hubbard Center for Genome Studies for the project Genomic Responses to the Deepwater Horizon event and development of high-throughput biological assays for oil spills. Additional support provided by the National Institutes of Health New Hampshire Idea Network of Biological Research Excellence (5 P20 GM 10350605).
The Gulf of Mexico Research Initiative (GoMRI) is a 10-year independent research program established to study the effect, and the potential associated impact, of hydrocarbon releases on the environment and public health, as well as to develop improved spill mitigation, oil detection, characterization and remediation technologies. An independent and academic 20-member Research Board makes the funding and research direction decisions to ensure the intellectual quality, effectiveness and academic independence of the GoMRI research. All research data, findings and publications will be made publicly available. The program was established through a $500 million financial commitment from BP. For more information, visit https://gulfresearchinitiative.org/.
© Copyright 2010-2019 Gulf of Mexico Research Initiative (GoMRI) – All Rights Reserved. Redistribution is encouraged with acknowledgement to the Gulf of Mexico Research Initiative (GoMRI). Please credit images and/or videos as done in each article. Questions? Contact web-content editor Nilde “Maggie” Dannreuther, Northern Gulf Institute, Mississippi State University (maggied@ngi.msstate.edu).




